กะโหลกศีรษะเชื่อมติดกันผิดปกติ (craniosynostosis)
นพ. นนท์ โรจน์วชิรนนท์
16 ส.ค. 2562
โรคที่มีความพิการในกลุ่มนี้ มีปัญหาจากการที่กระดูกที่มาประกอบกันเป็นกะโหลกศีรษะมีการเชื่อมติดกันผิดปกติ ทำให้กดรัดสมองไม่ให้เจริญขยายขนาดไปอย่างปกติ
รอยประสานกะโหลกศีรษะและใบหน้าที่มีการเชื่อมกันผิดปกติเกิดขึ้นขณะทารกอยู่ในครรภ์มารดา และเกิดต่อเนื่องภายหลังจากเกิดมาแล้ว โดยทั่วไปเราจึงสามารถวินิจฉัยโรคนี้ได้ตั้งแต่เกิด แต่มักจะไม่สามารถตรวจเห็นได้ด้วยเอกซเรย์อย่างอัสตราซาวน์ที่นิยมทำกันในช่วงตั้งครรภ์
โรคในกลุ่มนี้แบ่งออกได้เป็น 2 ประเภท คือ
- กลุ่มที่มีการเชื่อมติดของรอยประสานกะโหลกศีรษะหลายตำแหน่ง ร่วมกับความผิดปกติอวัยวะอื่นๆในร่างกาย (syndromic craniosynostosis) เช่น กลุ่มอาการครูซอง Crouzon Syndrome), กลุ่มอาการเอเพิร์ต (Apert Syndrome), กลุ่มอาการไฟเฟอร์ (Pfeiffer Syndrome), กลุ่มอาการมูนคู (Muenke syndrome), กลุ่มอาการเซเตอร์-โชเซน (Satre-Chotzen Syndrome), กลุ่มอาการคาร์เพนเตอร์ Carpenter Syndrome)
ผู้ป่วยเหล่านี้มีการเชื่อมติดของกระดูกกะโหลกศีรษะหลาย ๆ ชิ้น ทำให้มีกะโหลกศีรษะและใบหน้าผิดรูปอย่างรุนแรงไปพร้อม ๆ กัน ทั้งยังมีความพิการแต่กำเนิดของอวัยวะอื่น ๆ ของร่างกาย เช่น แขน ขา มือ เท้า หัวใจ กระดูกสันหลัง อวัยวะการได้ยิน กล้ามเนื้อตา ระบบต่อมไร้ท่อ - กลุ่มที่มีความผิดปกติเพียงรอยประสานกะโหลกศีรษะเพียงตำแหน่งเดียว (non-syndromic craniosynostosis) ทำให้บางด้านบางส่วนของกะโหลกศีรษะขยายตัวไม่ได้ ศีรษะมีรูปร่างบิดเบี้ยว เช่น ศีรษะยาวผิดปกติในแนวหน้าหลังคล้ายเรือ (scaphocephaly จาก sagittal craniosynostosis) ศีรษะรูปสามเหลี่ยม เป็นสันที่หน้าผาก (trigonocephaly จาก metopic craniosynostosis) ศีรษะเบี้ยว (plagiocephaly จาก unilateral coronal synostosis) ความผิดของกะโหลกจะนำมาซึ่งการผิดรูปของใบหน้า หากไม่ได้รับการรักษาโดยเร็วในวัยทารก
กลุ่มที่มีการเชื่อมติดของรอยประสานกะโหลกศีรษะหลายตำแหน่ง ร่วมกับความผิดปกติอวัยวะอื่นๆในร่างกาย (syndromic craniosynostosis)
ด้วยความก้าวหน้าทางการแพทย์โดยเฉพาะอย่างยิ่งในระดับโมเลกุลและพันธุศาสตร์ ทำให้ทราบว่าผู้ป่วยเหล่านี้ส่วนใหญ่มียีนที่ผิดปกติเป็นต้นเหตุ มีการกลายพันธุ์ของยีนที่ได้จากพ่อหรือแม่ในช่วงของการปฏิสนธิและตั้งครรภ์ ทำให้ผู้ป่วยส่วนใหญ่เป็นโรคขึ้นมาเป็นคนแรกในครอบครัว
ด้วยเหตุที่มียีนผิดปกติและยีนพวกนี้มีหน้าที่เกี่ยวกับกับการสร้างเนื้อเยื่อต่าง ๆ ในร่างกาย ทำให้ผู้ป่วยมักมีความผิดปกติของร่างกายในระบบต่าง ๆ ด้วย ไม่จำเพาะที่กะโหลกศีรษะ
และด้วยความรู้เรื่องยีนต้นเหตุที่มีมากขึ้น พบว่ามีโรคหลาย ๆ โรคเกิดจากการกลายพันธุ์ของยีนเดียวกัน จึงเริ่มมีการเรียกชื่อโรคตามยีนที่มีการกลายพันธุ์มากขึ้น เช่น FRFR2-related craniosynostosis หลาย ๆ โรคที่เคยวินิจฉัยไว้ ก็พบว่าผิดพลาดกลายเป็นอีกโรคหนึ่ง
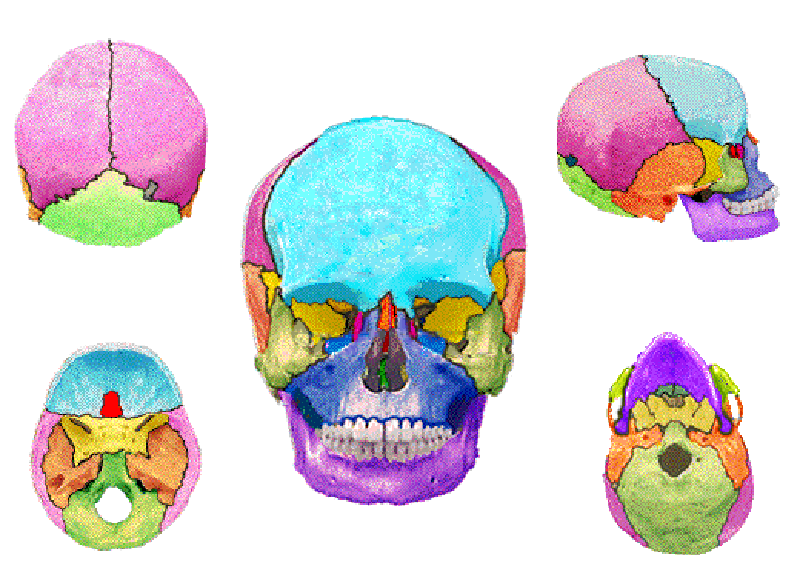
การเชื่อมติดผิดปกติของกระดูกกะโหลกศีรษะมีผลกับอวัยวะสามส่วน คือ
- การเชื่อมติดของรอยประสานกะโหลกศีรษะ (craniostenosis) - ทำให้การขยายขนาดกะโหลกศีรษะถูกจำกัด ทั้งยังมีลักษณะรูปร่างผิดไป เช่น มีลักษณะรูปทรงสูง (turricephaly) รูปทรงแหลมขึ้นไปตรงกลางศีรษะ (oxycephaly) หรือ แบนในแนวหน้าหลัง (brachycephaly) หรือบางครั้งรุนแรงจนศีรษะรูปร่างคล้ายดอกจิก (cloverleaf skull) การที่กะโหลกศีรษะไม่สามารถขยายตัวได้ตามปกติทำให้สมองซึ่งเจริญเติบโตอย่างรวดเร็วในช่วงอายุ 2-3 ขวบแรก ไม่สามารถขยายได้ตามปกติ อาจเป็นสาเหตุให้ผู้ป่วยมีปัญหาทางด้านพัฒนาการและสติปัญญาได้
- กระดูกเบ้าตาเชื่อมติดผิดปกติ (orbitostenosis) - กระบอกตาไม่สามารถขยายตามลูกตาที่โตขึ้น เป็นผลให้ลูกตาซึ่งมีขนาดใหญ่ขึ้นตามปกติไม่มีที่อยู่ จึงมองเห็นโปนมาทางด้านหน้า (exorbitism) จนบางครั้งหลับตาไม่สนิทหรือถลนหลุดออกนอกเบ้า จนเกิดเป็นแผลที่กระจกตา(ตาดำ)และตาบอดได้
- ใบหน้าส่วนกลางไม่สามารถขยายขนาด (faciostenosis) - ใบหน้าจึงมีลักษณะเว้าแบน ฟันบนอยู่หลังต่อฟันล่าง ประกอบกับบางรายมีเพดานที่สูงกว่าปกติ (high-arched palate) ทำให้โพรงจมูกเล็กลง หายใจลำบากจนขาดอ็อกซิเจนได้
ตัวอย่างโรคในกลุ่มนี้
กลุ่มอาการครูซอง (Crouzon syndrome)
อาการสำคัญในกลุ่มอาการครูซองคือ อาการตาโปนมาก บางรายโปนมากจนลูกตาถลนออกนอกเบ้า หลายรายมีตาเขเหล่ร่วมด้วย

กลุ่มอาการเอเพิร์ต (Apert syndrome)

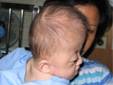


ตาจะโปนน้อยกว่าในกลุ่มอาการครูซอง แต่หน้าผากจะโปนนูนมากกว่า มีรอยคอดรัดเหนือคิ้ว และที่สำคัญมีนิ้วมือและนิ้วเท้าเชื่อมติดกันมาแต่กำเนิด
กลุ่มอาการครูซอง (Crouzon syndrome)
อาการสำคัญในกลุ่มอาการครูซองคือ อาการตาโปนมาก บางรายโปนมากจนลูกตาถลนออกนอกเบ้า หลายรายมีตาเขเหล่ร่วมด้วย
กลุ่มอาการไฟเฟอร์ (Pfeiffer syndrome)
มีความผิดปกติบริเวณใบหน้าและกะโหลกศีรษะที่คล้ายกลุ่มอาการเอเพิร์ต แต่มักมีหัวแม่มือและหัวแม่เท้าใหญ่กว่าปกติ และความผิดปกติของข้อศอกด้วย


กลุ่มอาการคาเพนเตอร์ (Carpenter syndrome)
นอกจากมีการเชื่อมของกะโหลกศีรษะก่อนกำหนดแล้ว จะมีความผิดปกติอื่นๆ เช่น มีนิ้วมือคด (clinodactyly) มือสั้น (short hand) หรือนิ้วเท้าบางนิ้วมีหนังเชื่อมติดกัน เป็นโรคชนิดเดียวในกลุ่มนี้ที่ถ่ายทอดทางกรรมพันธุ์ที่เป็นแบบยีนด้อย (autosomal recessive)
ตารางที่ 1 - Craniosynostosis syndromes ที่พบบ่อย
| ชื่อโรค | ชื่อรอยต่อกะโหลกที่เชื่อมเร็วผิดปกติ | อาการแสดง |
|---|---|---|
| ถ่ายทอดทางกรรมพันธุ์แบบยีนเด่น (autosomal dominant) | ||
| กลุ่มอาการครูซอง(Crouzon) | โคโรนอล (coronal), แซกจิตัล (sagittal) | กะโหลกศีรษะรูปร่างผิดปกติ ใบหน้าส่วนกลางไม่เจริญ ตาโปน กระบอกตาห่างกัน โพรงจมูกตีบแคบ |
| กลุ่มอาการเอเพิร์ต (Apert) | coronal, sagittal, แลมดอย (lambdoid), etc. | กะโหลกศีรษะรูปร่างผิดปกติ ใบหน้าส่วนกลางไม่เจริญ ตาโปน กระบอกตาห่างกัน ทางเดินหายใจแคบ นิ้วมือนิ้วเท้าติดกัน |
| กลุ่มอาการไฟเฟอร์(Pfeiffer) | ||
| 1. ชนิดที่ 1 | coronal, sagittal | กะโหลกศีรษะรูปร่างผิดปกติคล้ายกลุ่มอาการเอเฟิต แต่มีหัวแม่มือและแม่เท้าใหญ่ผิดปกติ นิ้วมือและเท้าอาจติดกันได้บ้างแต่ติดเฉพาะผิวหนัง |
| 2. ชนิดที่ 2 | ทุกรอยต่อ | เหมือนชนิดที่ 1 แต่โรครุนแรงกว่า ศีรษะเป็นรูปดอกจิก (cloverleaf หรือ Kleeblattschadel) ปัญญาอ่อน ข้อศอกติด |
| 3. ชนิดที่ 3 | ทุกรอยต่อและรุนแรงตั้งแต่เกิด | ตาโปนมาก กระบอกตาเล็กตื้น ใบหน้าส่วนกลางเว้าอย่างมาก ข้อศอกติดงอเหยียดไม่ได้ อายุสั้น |
| กลุ่มอาการเซเตอร์-โชเซ็น (Saethre–Chotzen) | coronal | กะโหลกศีรษะรูปร่างผิดปกติ มักจะแบนในแนวหน้าหลัง หนังตาตก อาจมีหนังของนิ้วติดกัน |
| ถ่ายทอดทางกรรมพันธุ์แบบยีนด้อย (autosomal recessive) | ||
| กลุ่มอาการคาเพนเตอร์(Carpenter) | ทุกรอยต่อ | ศีรษะรูปร่างผิดปกติ ร่วมกับมีจมูกแบน เอ็นหัวตาตก นิ้วเกิน |
ตารางที่ 2 - อุบัติการณ์และการถ่ายทอดทางกรรมพันธุ์
| ชื่อโรค | อุบัติการณ์ | ถ่ายทอดทางกรรมพันธุ์ | เป็นคนแรกในครอบครัว |
|---|---|---|---|
| Crouzon | 1:25,000 –40,000 | ยีนเด่น 67% | 33% |
| Apert | 1:50,000-100,000 | ยีนเด่น ส่วนน้อย | ส่วนใหญ่ พ่อมักมีอายุมาก |
| Carpenter | - | ยีนด้อย (โรคเดียวที่เป็น ยีนด้อย) | - |
| อื่นๆ | 1:25,000 | ใช่ | ใช่ |
การรักษา
ผู้ป่วยที่เป็นโรครอยต่อของกะโหลกศีรษะและใบหน้าเชื่อมเร็วกว่าปกติ (craniosynostosis) เช่น กลุ่มอาการครูซอง (Crouzon disease) กลุ่มอาการเอเพิร์ต (Apert syndrome) ฯลฯ ควรได้รับการผ่าตัดเพื่อขยายกะโหลกศีรษะและกระบอกตาในช่วงอายุก่อน 1 ขวบ คือ ระหว่าง 3-12 เดือน เพื่อให้มีพื้นที่ของสมองได้ขยายตัวและเจริญเติบโต รวมทั้งกระบอกตาเพื่อรักษาอาการตาโปน วิธีการนี้เราเรียกว่า การผ่าตัดทำ fronto-orbital advancement
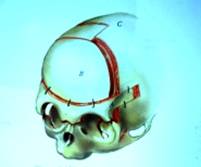
รูปที่ 5 – การทำผ่าตัดขยายกะโหลกศีรษะส่วนหน้าและเบ้าตา
ภายหลังจากนั้นต้องนัดผู้ป่วยมาดูเป็นระยะๆ ปีละครั้งเพื่อดูอาการว่า มีปัญหาแรงดันในสมองเพิ่มขึ้นหรือไม่ ตาโปนผิดปกติกลับขึ้นมาอีกหรือไม่ มีปัญหาการหายใจอันเนื่องมาจากใบหน้าส่วนกลางไม่เจริญเติบโตทำให้ทางเดินหายใจแคบหรือไม่ มีปัญหาเรื่องการเคี้ยวอาหารอันเนื่องมาจากการสบฟันที่ผิดปกติหรือไม่ เพื่อจะได้ดำเนินการแก้ปัญหาต่อไป
เมื่อเด็กโตขึ้นถึงระยะที่มีฟันแท้ขึ้นคือราวอายุ 10-12 ขวบ ทันตแพทย์จัดฟัน จะเข้ามามีบทบาทในการรักษาและจัดฟัน จากนั้นศัลยแพทย์อาจจะทำการยืดกระดูกใบหน้าส่วนกลางออกมาทางด้านหน้า โดยใช้เครื่องมือยืดกระดูก (distractor) ทำให้การหายใจและการสบฟันดีขึ้น



กลุ่มที่ผิดปกติที่รอยประสานกะโหลกศีรษะตำแหน่งเดียว (non-syndromic craniosynostosis)
หน้าผากและใบหน้าแบนยุบด้านใดด้านหนึ่ง (unilateral frontal plagiocephaly)
การที่ผู้ป่วยมีหน้าผากด้านใดด้านหนึ่งหรือทั้ง 2 ด้านยุบ, ไม่เจริญเติบโตออกมาด้านหน้าตามปกติเราเรียกลักษณะกะโหลกศีรษะแบบนี้ว่า "plagiocephaly" ถ้าเป็นข้างเดียวเราก็เรียก Unilateral โดยบอกไปว่าซ้ายหรือขวา ถ้าเป็นทั้งสองข้างเรียกว่า "bilateral" คำว่า plagios มาจากภาษากรีกแปลว่า ลาดเอียง, เฉียง (slant or oblique)
การที่หน้าผากแบนราบหรือบิดเบี้ยว อาจมีสาเหตุมาจากแรงภายนอก เช่น เด็กนอนทับอยู่ในท่านั้นนานๆ หรือ เกิดจากรอยเชื่อมของกะโหลกศีรษะ (coronal) ติดกันเร็วกว่าปกติ (synostosis) ซึ่งแพทย์จะต้องแยกให้ออก เพราะการรักษาต่างกัน กล่าวคือในรายที่เป็น unilateral coronal synostosis จะต้องให้การรักษาโดยการผ่าตัดเลื่อนกระดูกหน้าผากและกระบอกตาให้เลื่อนมาด้านหน้าให้เท่าข้างปกติในเด็กอายุ ก่อน 1 ขวบ เพราะถ้าทิ้งไว้จนโต ผู้ป่วยจะมีใบหน้าบิดเบี้ยว (facial distortion) ไปถึงกระบอกตา กรามบน กรามล่าง จมูก ทำให้การแก้ไขยุ่งยากมาก ในเวลาเด็กโตขึ้น กลุ่มพวกนี้จะพบได้ในเด็กแรกคลอดประมาณ 1: 10,000 แต่ถ้าในกรณีที่เกิดจากการนอนทับศีรษะในท่าเดียวนานๆ ก็อาจแนะนำวิธีแก้ไขท่านอนโดยไม่จำเป็นต้องผ่าตัดรักษาแต่อย่างใด
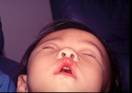
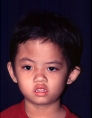
รูปที่ 6 – ผู้ป่วยศีรษะผิดรูปแบบ plagiocephaly